Protein folding has nothing to do with laundry. It is, in fact, one of the central questions in biochemistry. Protein folding is the continual and universal process whereby the long, coiled strings of amino acids that make up proteins in all living things fold into more complex three-dimensional structures. By understanding how proteins fold, and what structures they are likely to assume in their final form, researchers are then able to move closer to predicting their function.
This is important because incorrectly folded proteins in humans result in such devastating diseases as Alzheimer's, Parkinson's, Huntington's, emphysema and cystic fibrosis. Developing better modelling techniques for protein folding is crucial to creating more effective pharmaceutical treatments for these and other diseases.
Computational methods of modelling protein folding have existed for a couple of decades. But what McGill researcher Jérôme Waldispühl of the McGill Centre for Bioinformatics has done, working with collaborators from MIT, is to develop algorithms that can work from a laptop computer to examine a protein's fundamental chemical properties and then scan a number of possible protein shapes before predicting the final form that the protein is likely to take.
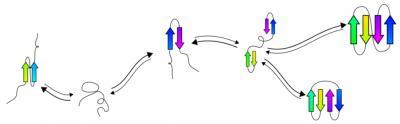
This graphic shows the process of predicting protein folding.
(Photo Credit: Tfolder)
The results have been impressive. Whereas classical techniques for predicting protein folding pathways required hundreds of thousands of CPU hours to compute the folding dynamics of 40 amino acids proteins, the program tFolder implemented by Solomon Shenker – a former McGill under-graduate student now at Cornell – has been able to predict correctly in 10 minutes on a single laptop, a coarse-grained representation of the folding pathways of a protein with 60 amino acids.
Waldispühl and his students continue to work on their algorithm to improve its success rate at predicting protein folding with broader categories of proteins including some that are important in DNA-binding. The research was recently presented at the 15th Annual International Conference in Research in Computational Molecular Biology (RECOMB 2011).
Source: McGill University