The research is funded by the National Institute of General Medical Sciences, National Institutes of Health.
Seeking new drugs that would allow chemotherapeutic compounds to enter and destroy cancer cells
Since the 1970s it has been known that the so-called multidrug resistance protein, P-gp, is most likely responsible for the failure of many chemotherapy drugs. P-gp is nature's way of pumping toxins from a cell, but if cancer cells express more P-gp than cells normally would, the chemotherapy is no longer effective because the protein considers it a toxin and pumps it out before it can destroy the cancer.
"We're looking for small molecules that will temporarily inhibit the pump; a new drug that could be co-administered with the chemotherapeutic and that stops the sump pump in the cancer cell so that the cancer chemotherapy can remain in the cell and kill the cancer," Wise said.
High-performance computer enables millions of digital screenings
Wise has run about 10.5 million computational hours since August 2009 and has screened roughly 8 million potential drugs against different protein structures.
"We are currently screening about 40,000 compounds per day on SMU's High Performance Computer," Wise said.
"We found a couple hundred compounds that were interesting, and so far we chose about 30 of those to screen in the lab," Vogel said. "From those, we found a handful of compounds that do inhibit the protein. We were thrilled. Now we're going back into the models and looking for other compounds that might be able to throw a stick in the pump's mechanism."
Massive increases in computational power in recent years have made the screening research possible, Wise said. "Ten years ago you couldn't have docked 8 million compounds — there just wasn't enough computational power."
Human P-gp: "We don't know what it looks like exactly."
Every organism has a version of P-gp. Its structure has been previously determined for some organisms — mostly bacteria, but also in mice — by studying the arrangement of atoms within protein crystals. However, the exact structure of the human enzyme remains unclear. Wise deduced the structure of human P-gp by relying on evolutionary relationships and scientific understanding of how proteins are put together. He then used computer programs to model the protein in a way that brings the static picture of the human pump to life in the computer. See http://bit.ly/Lncc6T.
To develop the model, Wise used freely available simulation software developed by researchers at the University of Illinois, the National Institutes of Health and the Scripps Research Institute. Wise and Vogel use compounds from ZINC, a free database of more than 21 million commercially available compounds for virtual screening. ZINC is provided by the Department of Pharmaceutical Chemistry at the University of California, San Francisco.
"We can physically build these molecules in the computer, in silico, and computationally we can model a variety of conditions: We can raise the temperature to 37 degrees Centigrade, we can have the right salts and all the right conditions, just like in a wet-lab experiment. We can watch them thermally move and we can watch them relax," Wise said. "The software is good enough that the model will move according to the laws of physics and the principles of biochemistry. In this way we can see how these compounds interact with the protein in a dynamic way, not just in a snapshot way."
Even with the 3D dynamic model and a supercomputer, the odds are stiff
Theoretically, if a drug can be found that can temporarily knock out the sump-pump proteins, then all those cancer chemotherapies that don't work for a patient will work again.
"The ultimate goal of our research would be to find a compound that is safe and effective," Wise said. Even with a supercomputer, however, the odds are steep.
"Out of a hundred good inhibitors that we might find, 99 of them might be extremely toxic and can't be used. In the pharmaceutical industry there are many, many candidates that fall by the wayside for one reason or another," he said. "They metabolize too quickly, or they're too toxic, or they're not soluble enough in the acceptable solvents for humans. There are many different reasons why a drug can fail. Finding a handful has been a great confirmation that we're on the right track, but I would be totally amazed if one of the first we've tested was the one we're looking for."
Vogel is an associate professor and director of SMU's Center for Drug Discovery, Design and Delivery. CD4 was launched by SMU's Biological Sciences and Chemistry departments and has as its mission the search for new drug therapies and delivery methods that can be developed into clinical applications.
SMU biologists Pia Vogel and John Wise are using the computational power of SMU's high-performance supercomputer to screen millions of drug compounds in hopes of finding one that will aid in the fight against recurring cancer.
Wise has deduced what is believed to be the first dynamic model of the human P-glycoprotein, known for its ability to pump out chemotherapy from a cancer cell, allowing the cell to survive.
Using the dynamic model, Wise and Vogel have found some compounds that "bind" to the protein. This provides hope they can find a compound that will throw a wrench into the cell's ability to pump out cancer-killing chemotherapy by disabling the energy source that powers the pump. Video summary, http://bit.ly/MeUFOo.
(Photo Credit: www.smuresearch.com)

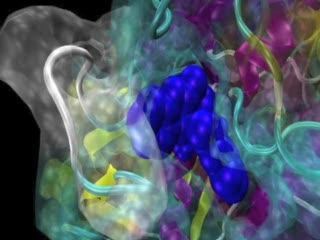
Purple and blue helices, yellow ribbons and turquoise coils form the backbone skeletal structure of the human multi-drug resistance protein. A newly identified inhibitor binds to one of the pumps two ATP binding sites (dark blue). In motion at http://bit.ly/Lncc6T.
(Photo Credit: John Wise, SMU)
Source: Southern Methodist University